
Descriptive Analysis of MLST Data for MRSA
Want to share your content on R-bloggers? click here if you have a blog, or here if you don't.
During one of my summers, I had the opportunity to conduct some research on the prevalence of methicillin-resistant Staphylococcus aureus (MRSA) in vulnerable populations and examining US emergency department data and I thought this would be a pretty interesting topic to expand on for my thesis in lieu of the increasing concerns of antimicrobial resistance, both at the domestic and international level. This resulted in an systematic review of the recent literature of MRSA in the major Far East countries (China, Hong Kong, Japan, South Korea and Taiwan) which has a wide variety of genotypes and strains. If you’re interested in understanding S. aureus in Asia, a good starting point is this article. (An extensive, yet most likely the best review on community-associated MRSA written by two researchers from Chicago if you really want to dig deep into this topic).
What is MRSA?
MRSA is a gram-positive bacteria which is found on almost every continent on earth, which developed resistance to methicillin (a narrow spectrum β-lactam antibiotic from the penicillin class) in the early 60s. MRSA was originally considered to only cause infections within hospital settings (ICUs, EDs, surgery) but towards the end of the 1980s and into the 1990s, cases of severe MRSA infections were being reported in communities outside of healthcare centers. MRSA typically causes skin and soft tissue infections but some cases when left untreated can cause havoc to respiratory system, bloodstream and eventually cause sepsis – not a pretty picture. Over the next 20 years, reports of different strains of this “new” MRSA started to appear around the globe. Some of these strains successfully disseminated from their country of origin to neighboring regions and even across continents with the help of globalization. With the advent and advancement of genotyping technology – researchers were able to track and identify the strains causing outbreaks across the world. One classification system is called Multi Locus Sequence Typing (MLST) which was developed by researchers at Imperial College, UK. In short, it is based on creating a profile or sequence type (ST) of each S. aureus strain based on sequencing specific internal nucleotide fragments of seven house-keeping genes. Using this method, investigators are able to discriminate between strains and identify evolutionary changes over time.
Reading in the data using R
This post has nothing too fancy in R – I’m just going to do a step-by-step walkthrough of data cleaning and asking a couple questions, then running descriptive analyses to find some answers. For those you are proficient in doing this might find this a little boring (you can just scroll down to the end and see the results). The data is taken from http://saureus.mlst.net/ by selecting the link “download as MS excel” and downloading the spreadsheet to a local directory. The data is complied from submissions to the database which report new MLSTs with some demographic information. This database doesn’t really provide information of a specific strain’s prevalence – it’s just a timestamp report for novel strains. This file contains nine sheets. The one using for this post is called “profile”. I saved this as a comma-delimited file manually but there are lots of resources on the internet of doing this in R using some packages. If you’re interested take a look at this or this – I’ve tended to use the readxl package developed by Hadley Wickham when I’m working with a lot of .xls files.
So now you should have a .csv file someone on your local directory. You first want to set your working directory. You can read a brief description of how to do this here. I’m using R 3.3.2 on Mac OS as an FYI, the link before talks about the differences when you set your working directory in Windows and Mac OS.
getwd() #shows what your current working directory is
setwd("/Users/myname/Documents/Folder") #sets your working directory
Next, we’re gonna read the comma delimited file into R using the read.csv() function and assigning it the name “profile”. The first argument you want input the filename with the .csv extension. The second argument tells the function that the first row in the file contains the headers or column names. Based on a quick visual inspection of spreadsheet file using Excel, there are many missing values just left as blank cells. The final argument I used specifies to convert any blank cells to NA type so that R can just treat these as missing data.
profile<-read.csv("name_of_your_file.csv",header=T,na.strings=c("",NA))
A good practice I always do when I read in spreadsheets into R is to get a idea of what I’m dealing with. I want to know how many columns and rows there are, the names of the headers/columns and the type of data in each column. To accomplish this, we run the dim(), names() and str() functions for each respective question.
dim(profile) #dimensions of the dataset names(profile) # names of the columns in dataset str(profile) #structure of the data frame
When we do this, the data frame should have 4703 rows and 25 columns. The 25 headers include the following…
[1] "id" "strain" "other_name" "st" "country" [6] "region" "year" "age_yr" "age_mth" "sex" [11] "disease" "sourcce" "epidemiology" "species" "oxsau" [16] "methicillin" "vancomycin" "spa_type" "reference" "comments" [21] "sender" "curator" "date_entered" "datestamp" "username"
Most of the time the columns names can be cryptic. You always want to have the metadata or the documentation files for the datasets so that you know exactly what each column represents. In this case for this file, it is pretty straightforward – we can see that for each submission there is an id number, strain name, sequence type (st), who submitted the info, time/date stamps and some other demographic information.
When we run str(profile), our output shows the class type of each column. This will be important as R tends to have functions that run only for specific types of data. A good overview the different types of data in R can be found here.
Two major things stick out to me when I look through the data – first the column with country names is type factor. I want to convert into a string type, so later on I can subset the data by country by referring to the actual name, versus the factor level (which would be confusing and annoying to keep track of).
# Change country to character from factor type profile$country <- as.character(profile$country)
Second, I noticed the classification of a lot of categorical columns have multiple levels. Sex has 6 levels!? Methicillin resistance has 9 levels? What does this mean…? Well, the problem for databases that are derived from public submissions is that there is no standardization with the data input. When we inspect what the levels are for sex by running the line of code below, we see that due to the lack of data input standardization, we have F, female and Female for those categorized as female sex. For male sex we have M and Male, and then Unspecified for those contributors who didn’t leave it blank.
> summary(profile$sex) F female Female M Male Unspecified NA's 17 1 814 19 842 1343 1667
I’m not particularly interested in distribution of gender with these reports so I’m okay with leaving this alone. But we have to same issue with the column for methicillin resistance and I want to simplify it to a binary category of resistant and susceptible. When we look at the levels for the the methicillin column we see this…
> summary(profile$methicillin) I MIC 4mg/L MIC 64mg/L r R s S 1 2 1 1 1533 3 2175 Unknown Unspecified NA's 4 199 784
Since we want to simplify this, we can use the levels() function to set and rename the levels in this column. First, we’re gonna merge the lower and uppercase letters together (r will be R and s will be S)
levels(profile$methicillin) <- c("I","MIC4","MIC64","R","R","S","S","Unknown","Unspecified")
Minimum Inhibitory Concentration (MIC) is the lowest concentration of an antibiotic that inhibits the growth of an organism. If you have two strains of S. aureus (#1 and #2) where #1 has a higher MIC to methicillin or oxacillin (another antibiotic) compared to strain #2 strain, #1 is considered to be more resistant to that tested antibiotic compared to #2. This is a method used to provide more granularity in defining antimicrobial resistance. Here in this case, I recognize MIC’s above 4mg/L are considered resistant (I’m making an assumption that all the reports used the same standard and technique to make this definition, a limitation is simplifying this). There’s more information on MIC’s for MRSA on the CDC’s website. So with all this in mind, I going to merge the MIC4 and MIC64 into the resistant category.
levels(profile$methicillin) <- c("R","R","R","R","S","Unknown","Unspecified")
After doing this, we only have four levels in the methicillin column: R, S, Unknown and Unspecified. We can check this by running
factor(profile$methicillin)
Before we move on, another good practice is to be aware (and quantify) missing values in each of the columns. Lets say we want to know how many missing sequence types (STs) are there? We can use the is.na() and which() together to figure out which rows are missing values under the st column.
> which(is.na(profile$st)) [1] 1596 1897 4602 4605
Using this, we now know that there are four rows missing STs. Row numbers 1596, 1897, 4602 and 4605. If we wanted to subset the data for those missing STs, we can just use square brackets “[]” to accomplish this. If you’re unfamiliar with this, you can read this post to learn more on these accessors.
profile[which(is.na(profile$st)),]
Let’s ask some questions
Now that we’re done with the appropriate cleaning of the data, let’s ask some (random) questions
1. Which countries reported sequence type 239 (ST-239)?
Based my literature review, ST-239 is a common hospital-associated strain in Asia and it’s prevalence has increased in China, where is it the most common strain in the entire country. Other variants of ST-239 are common in Eastern Europe and South America as well, in particular Hungary and Brazil respectively.
To answer this question, we use the logic statements with subsetting to accomplish this.
id.239 <- which(profile$st==239) #index of rows with ST-239 st.239 <-profile[id.239,] #new dataframe of only ST-239 sort(unique(st.239$country)) # alphabetical list of countries with ST-239
When we run the sort() function we get a vector of 29 countries in alphabetical order which have reported ST-239.
[1] "Algeria" "Australia" "Brazil" [4] "China" "Czech republic" "Eire" [7] "Finland" "Germany" "Greece" [10] "Hungary" "India" "Iran" [13] "Malaysia" "Pakistan" "Poland" [16] "Portugal" "Scotland" "Slovenia" [19] "South Africa" "Spain" "Sweden" [22] "Switzerland" "Taiwan" "Thailand" [25] "The Netherlands" "Turkey" "UK" [28] "United Arab Emirates" "USA"
2. Which countries reported ST8 and spa type t008?
spa typing is another classification technique used for S. aureus strains. It is a single-locus typing method, so it’s much more cost-effective compared to MLST.
We use the similar code as above except we add another condition to the logic statement to specify the spa type.
id.ST8.t008 <- which(profile$st==8 & profile$spa_type=="t008") ST8.t008 <- profile[id.ST8.t008,] sort(unique(ST8.t008$country))
We see that 4 countries that have reported ST-8 with spa type t008.
[1] "Algeria" "France" "Portugal" "Switzerland"
3. What is the proportion of methicillin-susceptible vs. -resistant S. aureus reports are there in North America?
I was honestly just curious, the results we get from this are definitely limited due to the assumptions we had with the definitions and simplification discussed earlier.
First, since there is no column specifying the region of North America, we have to specify which countries to include to this subset. By visual inspection of the countries using sort(unique(profile$country)), we see that there Canada and USA but no Mexico. So we’ll subset the data to only Canada and USA.
NorthA.MRSA <- profile[profile$country=="USA" | profile$country=="Canada",] #selection of North American countries
Then running the following line, we get the counts of methicillin-resistant and methicillin-susceptible in North America.
table(NorthA.MRSA$methicillin,useNA="always") R S Unknown Unspecified <NA> 138 148 0 15 75
4. How many different STs are there in China, S Korea, Taiwan and Japan?
This is just coming from my thesis and getting an idea of distributions in the region of the world.
First we subset the data so that it only includes the countries of interest…
F.east <- profile[profile$country=="China" | profile$country=="South Korea" | profile$country=="Japan" | profile$country=="Taiwan",]
The following lines of code will answer some other questions related to this…
factor(F.east$st) #find the number of STs in all these countries sum(is.na(F.east$st)) #number of missing values table(F.east$country) #counts of STs by country
Based on 573 entries, we find that 289 different STs; China has 241, Japan has 259, South Korea has 26 and Taiwan 59.
If we want to only look those without missing values…
FE.final<-F.east[complete.cases(F.east$country),] #removal of all rows that are missing an entry for country
To go further we can look at each specific country, lets ask what are the top 5 most reported STs in China?
FE.China <- FE.final[FE.final$country=="China",] #subset for China sort(table(FE.China$st),decreasing=T)[1:5] #top 5 most reported > sort(table(FE.China$st),decreasing=T)[1:10] #top 10 most frequent 97 239 398 88 2154 21 13 5 3 3
5. What are the 10 most frequently STs reported in North America in this database?
Let’s also visualize this in a basic pie chart? First need to setup the data so that it is easier to visualize using the ggplot2 package. So we setup the STs for North America and then order the the counts/frequencies of STs. *note you can also accomplish this using the count() function in plyr package
c.NorthA.MRSA <-as.data.frame(table(NorthA.MRSA$st))
names(c.NorthA.MRSA) <- c("ST","count") #data frame containing count frequencies of the STs
c.NorthA.MRSA<-c.NorthA.MRSA[order(-c.NorthA.MRSA$count),] #order the STs by count frequencies
c.NorthA.MRSA[1:10,] # top ten
Then we use ggplot2 to create the pie chart! First specifying the theme so we can tinker around with font size, remove the background grid, etc. I used the scales package to display the percentages in the pie chart for each ST. I stumbled across this incredible resource for color palettes for data visualization where you can choose distinct colors (with color-blind friendly options as well!). This specific case I choose the pastel color option.
blank_t <-theme(
axis.title.x = element_blank(),
axis.title.y = element_blank(),
panel.border = element_blank(),
panel.grid=element_blank(),
axis.ticks = element_blank(),
plot.title=element_text(size=20, face="bold",hjust=0.6),
plot.subtitle=element_text(size=15,face=c("bold","italic"),hjust=0.6),
axis.text.x=element_blank(),
legend.title=element_text(size=14,face="bold",vjust=0.5),
panel.margin=unit(2,"cm")
)
pie1<-ggplot(c.NorthA.MRSA[1:10,], aes(x="",y=count,fill=ST,order=ST)) +
geom_bar(width=1, stat="identity") +
ggtitle(element_text(size=50))+
labs(title = "STs in North America", subtitle = "Canada and United States",y=NULL) +
coord_polar(theta="y")+
geom_text(aes(label = scales::percent(count/sum(c.NorthA.MRSA[1:10,2]))),size=4, position = position_stack(vjust = 0.6)) +
blank_t +
scale_fill_manual(guide_legend(title="STs"),
values=c("#e6b4c9",
"#bce5c2", # color palette hexcodes
"#c7b5de",
"#e3e4c5",
"#a1bbdd",
"#e5b6a5",
"#99d5e5",
"#bdbda0",
"#dcd1e1",
"#99c6b8"))
print(pie1)
ggsave(plot=pie1,filename="North_America_STs_MLSTdatabase.png", width=5.75, height=7, dpi=120)
Then we get the pie chart that looks like this…
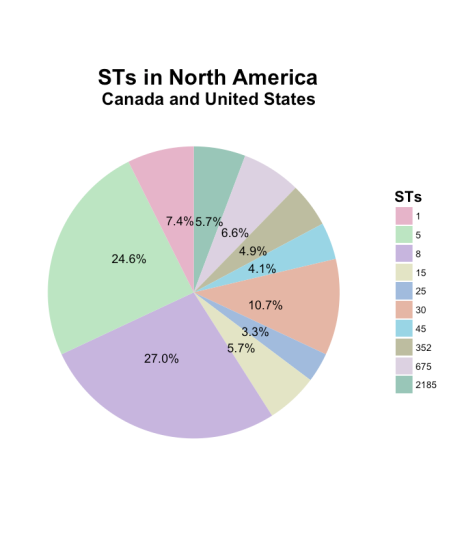
We can see that ST-5 and ST-8 have the most number of reports, while the remainder are ≤10%. All the code is on Github, open for anyone to use and adapt with. I’m always open for suggestions to how to improve the methodology and code. I know pie charts are “controversial” so any input on other graphics to display this type of information is always welcome.
Hopefully this post demonstrates the power and flexibility of R when you some questions and how easily you can find the answers in your data. It is always important to remember where the data came from, who collected it, when you received it, how you processed it and any assumptions you make along the way – this can change the interpretation for the results dramatically. The results from this specific dataset are just informative and descriptive of the MLST database and potentially lead to some hypotheses. Another possible extension of this data is attaching the strain information by country and displaying it in a map. The MLST database website for S. aureus already has done this with Google Maps (click the link MLST-Maps in the top menu). There is a wealth of resources on mapping in R (the link is from the R-Bloggers website) and many packages exist for displaying spatial data – so this might be something I might tinker with in the near future.

R-bloggers.com offers daily e-mail updates about R news and tutorials about learning R and many other topics. Click here if you're looking to post or find an R/data-science job.
Want to share your content on R-bloggers? click here if you have a blog, or here if you don't.