

Want to share your content on R-bloggers? click here if you have a blog, or here if you don't.
*Contributed by Vinicius Bastazini and Vanderlei Debastiani
“Nothing in biology makes sense except in the light of evolution.” (Dobzhansky 1973)
“Nothing in evolutionary biology makes sense except in the light of ecology.” (Grant and Grant 2008)
“Nothing in evolution or ecology makes sense except in the light of the other. ” (Pelletier et al. 2009)
Understanding the interplay between evolutionary and ecological processes is crucial for describing patterns of species diversity in space and time. Historically, community ecology and evolutionary biology have followed separate paths. However, in the past decades, the inclusion of phylogenetic data has expanded the toolkit for community ecologists, shedding light on large range of biological processes such as species interactions, community assembly and disassembly, species distribution, and ecosystem functioning. Phylogenetic information has also provided a sort of “temporary” solution for missing data in ecological databases.
This novel and integrative approach has given rise to Ecophylogenetics, an interdisciplinary field of study that combines principles from both ecology and phylogenetics. It focuses on understanding the evolutionary relationships among species in the context of ecological processes and patterns.
In this context, phylogenetically structured models have thus revolutionized our understanding of ecological phenomena, by providing a framework to explore and interpret the patterns of species evolution, diversity, and interactions. This is especially relevant in Network Ecology, a branch of ecology interested in investigating the structure, function, and evolution of ecological systems using network models and analyses. Phylogenetic approaches have been incorporated into the study of ecological networks formed by interacting species, in order to describe distinct ecological phenomena, such as network assembly and disassembly (see a related post here), and its ability to cope with cascading effects such as species loss (see a related post here).

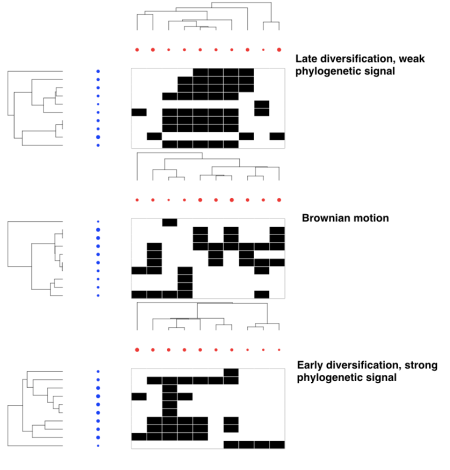
Examples of some of the possible eco-evolutionary dynamics of bipartite mutualistic networks. Species interactions are represented by black cells in the bi-adjacency matrices along with their evolutionary trees and traits ( blue and red circles). Trait values are represented by circle size.
In the following example, we will demonstrate how to simulate phylogenetically structured networks using R.
In this example we will simulate ultrametric phylogenetic trees for two distinct sets of species, such as consumers ( e.g., predator, pollinator or seed disperser) and resources (prey, flowering plants), following a uniform birth–death process, which reflects the underlying evolutionary dynamics of taxa. Additionally, we will simulate the evolution of a single trait by employing a family of power transformations to the branch lengths of these simulated phylogenetic trees. The choice of this transformation parameter defines the tempo and mode of trait evolution. Networks of interacting species will be generated using a single-trait complementarity model (Santamaria and Rodriguez-Girones 2007). According to the single-trait complementarity model, each species in the network is represented by a mean trait value and its variability, and a pair of species is more likely to interact if their trait values overlap. You can find further information on this sort of simulation in Debastiani et al. (2021a, 2021 b) and Bastazini et al. (2022).
#### Load R packages
require(ape)
require(geiger)
require(plotrix)
#### Define phylo_web, a function that generates phylogenetic structured networks
# The inputs are: number of species in each set of interacting species and the level of phylogenetic signal
phylo_web = function(n_spe_H, n_spe_L, power_H, power_L) {
# Generate a phylogenetic tree for consumer species (H)
tree_H = geiger::sim.bdtree(b = 0.1, d = 0, stop = "taxa", n = n_spe_H, extinct = FALSE)
for (y in 1:length(tree_H$edge.length)) {
if (tree_H$edge.length[y] <= 0)
tree_H$edge.length[y] = 0.01
}
# Generate a phylogenetic tree for resource species (L)
tree_L = geiger::sim.bdtree(b = 0.1, d = 0, stop = "taxa", n = n_spe_L, extinct = FALSE)
for (y in 1: length(tree_L$edge.length)) {
if (tree_L$edge.length[y] <= 0)
tree_L$edge.length[y] = 0.01
}
# Assign labels to the tips of the trees
tree_H$tip.label = sprintf("H_%.3d", 1:length(tree_H$tip.label))
tree_L$tip.label = sprintf("L_%.3d", 1:length(tree_L$tip.label))
# Generate trait data for consumers
trait_H = matrix(NA, nrow = n_spe_H, ncol = 1)
for(n in 1:1) {
trait_H[, n] = ape::rTraitCont(compute.brlen(tree_H, power = power_H), model = "BM")
}
trait_H[,1] = plotrix::rescale(trait_H[,1], newrange = c(0, 1))
rownames(trait_H) = tree_H$tip.label
colnames(trait_H) = sprintf("Tr_H_%.3d", 1:ncol(trait_H))
# Generate trait data for resources
trait_L = matrix(NA, nrow = n_spe_L, ncol = 1)
for(n in 1:1) {
trait_L[, n] = ape::rTraitCont(compute.brlen(tree_L, power = power_L), model = "BM")
}
trait_L[,1] = plotrix::rescale(trait_L[,1], newrange = c(0, 1))
rownames(trait_L) = tree_L$tip.label
colnames(trait_L) = sprintf("Tr_L_%.3d", 1:ncol(trait_L))
# Generate bi-adjacency interaction matrix based on trait differences and niche overlap
d_H = matrix(runif(n_spe_H, max = 0.25), nrow = n_spe_H, ncol = 1)
d_L = matrix(runif(n_spe_L, max = 0.25), nrow = n_spe_L, ncol = 1)
web = matrix(NA, nrow = n_spe_L, ncol = n_spe_H)
for(i in 1:n_spe_L) {
for(j in 1:n_spe_H) {
II = abs(trait_H[j] - trait_L[i])
III = 0.5 * (d_H[j] + d_L[i])
web[i,j] = ifelse(II < III, yes = 1, no = 0)
}
}
colnames(web) = tree_H$tip.label
rownames(web) = tree_L$tip.label
# Ensure there are no species with no interactions
z_H = which(colSums(web) == 0)
if (length(z_H) > 0) {
for(i in 1:length(z_H)) {
web[sample(1:n_spe_L, size = 1), z_H[i]] = 1
}
}
z_L = which(rowSums(web) == 0)
if (length(z_L) > 0) {
for(i in 1:length(z_L)) {
web[z_L[i], sample(1:n_spe_H, size = 1)] = 1
}
}
# Return the results as a list
RES = list(tree_H = tree_H, tree_L = tree_L, trait_H = trait_H, trait_L = trait_L, web = web)
return(RES)
}
#### Generate networks using the phylo_web function
# Here, we are generating a network formed by 10 resource species (e.g., flowers) , 10 consumers (pollinators), simulating a Brownian motion process for trait evolution:
n_spe_HA = 10 # Number of consumer species
n_spe_LA = 10 # Number of resource species
power_HA = 1 # Grafen's rho of consumer species
power_LA = 1 # Grafen's rho of resource species
web = phylo_web(n_spe_HA, n_spe_LA, power_HA, power_LA)
# The result is list containing a bi-adjacency matrix for two sets of interacting species, their phylogenies and trait values.
web
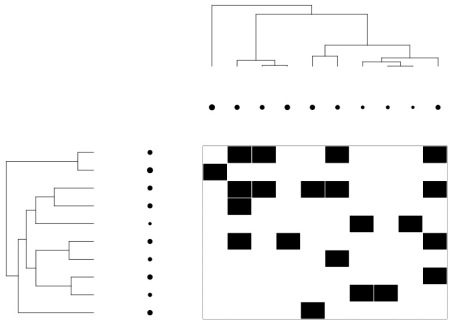
Now we can plot the bi-adjacency matrix along with their evolutionary trees and traits. Trait values are represented by circle size.
# Create and arrange the plot for the phylogenetic trees, species traits, and the network
par(mar = c(0.4, 0.4, 0.4, 0.4))
layout(matrix(c(0, 0, 1, 0, 0, 2, 3, 4, 5), nrow = 3, ncol = 3, byrow = TRUE),
widths = c(0.4, 0.4, 1),
heights = c(0.4, 0.4, 1))
# Plot
ape::plot.phylo(web$tree_H, show.tip.label = FALSE,
direction = "downwards")
plot(web$trait_H[,1], rep(1, times = length(web$trait_H[,1])),
xlab = "", ylab = "",
xaxt = "n", yaxt = "n",
type = "n", bty = "n",
ylim = c(0.9, 1.1), xlim = c(1, length(web$trait_H[,1])))
points(1:length(web$trait_H[,1]), rep(1, times = length(web$trait_H[,1])),
cex = web$trait_H[,1]+1, pch = 19)
ape::plot.phylo(web$tree_L, show.tip.label = FALSE,
direction = "rightwards")
plot(web$trait_L[,1], rep(1, times = length(web$trait_L[,1])),
xlab = "", ylab = "",
xaxt = "n", yaxt = "n",
type = "n", bty = "n",
xlim = c(0.9,1.1), ylim = c(1, length(web$trait_L[,1])))
points(rep(1, times = length(web$trait_L[,1])), 1:length(web$trait_L[,1]),
cex = web$trait_L[,1]+1, pch = 19)
plotrix::color2D.matplot(web$web,
cs1 = c(1, 0), cs2 = c(1, 0), cs3 = c(1, 0),
yrev = FALSE,
ylab = "", xlab = "",
xaxt = "n", yaxt = "n",
border="white", axes = FALSE)
Et voilà!!!

R-bloggers.com offers daily e-mail updates about R news and tutorials about learning R and many other topics. Click here if you're looking to post or find an R/data-science job.
Want to share your content on R-bloggers? click here if you have a blog, or here if you don't.